
Abstract
Introduction
Acute pain continues to be a health burden worldwide [1]. Multimodal analgesia via a combination of two or more analgesic medications with different mechanisms of action that work additively has the potential to improve analgesia while reducing the dose and side effects of any single pain medication [2]. The American Society of Anesthesiologists recommends a multimodal approach, whenever possible, for the management of acute pain following surgery [3]. In addition to nonsteroidal anti-inflammatory drugs (NSAIDs), opioids can be administered due to their different mechanism of action, which targets other pain pathways, resulting in potential additive effects [3].
Diclofenac is one of the most widely used NSAIDs, with proven efficacy as an analgesic for acute postoperative pain [4]. The analgesic and anti-inflammatory effects of diclofenac are mediated by inhibition of the two main isoforms of cyclooxygenase (COX-1 and COX-2), an enzyme involved in the biosynthesis of prostaglandin from arachidonic acid. Diclofenac has a higher selectivity for the COX-2 isoform, but its plasma concentration undergoes wide fluctuations, with the consequence that it inhibits both COX isoforms equally at the beginning of the dosing interval. The selectivity of diclofenac for COX-2 only becomes apparent when plasma levels drop [5]. Prostaglandins are important mediators of hyperalgesia caused by tissue damage through inflammation, trauma, or edema. Inhibition of COX-2 and, consequently, prostaglandin E2 production in inflamed tissue and in the dorsal horn of the spinal cord downregulates hyperalgesia and reduces pain [6-8]. However, prostaglandins are also involved in other functions such as tissue protection in the gastrointestinal tract, increasing the osmotic resistance of kidney tubular cells and regulation of blood coagulation. Therefore, the long-term use of diclofenac is also associated with an increased risk of gastrointestinal, renal, and cardiovascular adverse events. The general recommendations to minimize the risk of serious adverse events are to use diclofenac at the lowest effective dose for the shortest duration of time [9-11].
Tramadol is a weak opioid with analgesic potency 1/5 to 1/20 that of morphine [12], which exerts its analgesic effect via at least two complementary mechanisms: binding of the parent compound and M1 metabolite to μ-opioid receptors and weak inhibition of the re-uptake of norepinephrine and serotonin [13]. Tramadol is effective and recommended for the treatment of moderate to severe pain [14]. Long-term use of tramadol can be associated with tolerance and dependence, often requiring careful and regular monitoring; however, the most frequent adverse events (AEs) associated with the short-term use of tramadol (the setting for the current study) include nausea, vomiting, and dizziness [14].
Preclinical studies undertaken to analyze the possible benefits of combined therapy with tramadol and diclofenac found synergistic interactions between these analgesics in thermal hyperalgesia, hot plate and hot water tail immersion models of nociception in rats, suggesting good therapeutic potential of this combination in the treatment of acute and inflammatory pain [15, 16].
Further, a pharmacokinetic interaction study (DCLF/TRMD-GRNN-01; data on file) showed that the extent of absorption of tramadol and diclofenac with the fixed-dose combination (FDC) was bioequivalent to that with either monotherapy and that there was no clinically significant pharmacokinetic interaction between tramadol and diclofenac, indicating that the FDC was acceptable from a pharmacokinetic perspective.
In the clinical setting, several early studies in subjects undergoing surgeries, including lumbar disc surgery [17], cesarean delivery [18-20], and abdominal surgeries [21], showed that coadministration of tramadol and diclofenac (administered parenterally and/or rectally) significantly reduced the intensity of pain [17-19] and produced rapid analgesia [20, 21]. In addition, a pilot study in 30 patients showed that an oral FDC of tramadol and diclofenac provided significant pain relief at three and six hours postdose and was generally well tolerated in patients undergoing orthopedic surgery [22].
The present phase 3 trial (ClinicalTrials.gov NCT number NCT03714672) aimed to assess the efficacy and safety of an FDC of tramadol hydrochloride and diclofenac sodium in two strengths (25 mg/25 mg and 50 mg/50 mg) using a postoperative dental pain model because of its assay sensitivity. The Dental Impaction Pain Model has been used since 1975 to simplify and accelerate analgesic drug development [23, 24]. It provides a valid and efficient method of assessment with high assay sensitivity and is widely used for early phase 2 and regulatory trials [24]. The bilayered FDC tablet consists of an immediate-release tramadol layer providing rapid-onset analgesia and a sustained-release diclofenac layer providing more prolonged analgesia. A novelty of this short-term dental impaction study was the noninferiority study design.
Methods
Patients
Eligible subjects were >18 to 60 years of age who were in good general health and required extraction of three or more third molars with two mandibular partial or full bony impacted third molars. Subjects were excluded if they had molars close to the mandibular canal, required immediate dental procedures other than extraction of the third molars, had a history of seizures, known alcohol or drug abuse in the last six months, or hypersensitivity to any of the investigational medicinal products (IMPs) or the anesthetic used during surgery or the rescue medication (ibuprofen, ketorolac). Also excluded were subjects who received >300 mg of lidocaine in total, a long-acting NSAID within 24 hours, or five times the elimination half-life of that NSAID (whichever was longer) before surgery, any systemic corticosteroid, any analgesic medication other than short-acting preoperative or intraoperative anesthetic agents within 24 hours before taking IMPs, or an analgesic medication other than the IMPs immediately after the surgical procedure. Subjects who did not achieve pain intensity of ≥5 points on an 11-point numerical rating scale (NRS; score range 0-10) within four hours after the surgical procedure were ineligible for the study.
Study Design
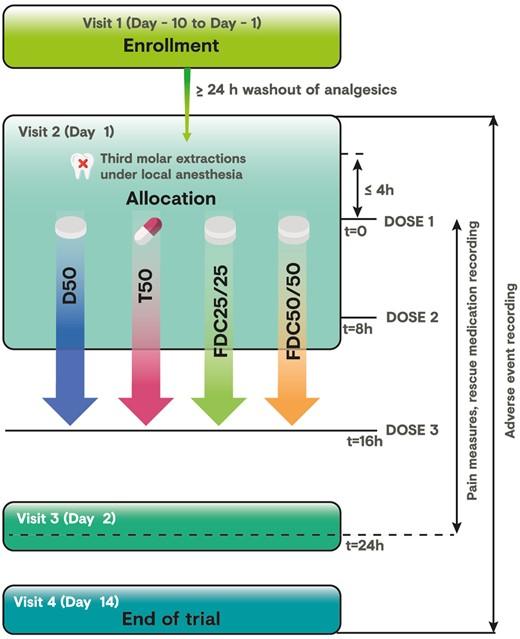
This was a prospective, randomized, double-blind, multisite, diclofenac- and tramadol-controlled, parallel-group, phase 3 trial to compare the analgesic efficacy and safety of two doses of tramadol HCl/diclofenac sodium FDC 25 mg/25 mg (FDC 25/25) and 50 mg/50 mg (FDC 50/50) vs tramadol 50 mg (T50) and diclofenac 50 mg (D50) monotherapies in subjects with moderate to severe acute pain after third molar tooth extraction. Eligible subjects received three doses of blinded study medication at eight-hour intervals over a 24-hour period. The first dose was administered at the research site, and the second dose was administered eight hours (±10 minutes) later, after which subjects were allowed to leave the study site. Subjects took the last dose of study medication at home, 16 hours (±1 hour) after the first dose was administered. The trial was performed at eight sites across Mexico and conducted following local guidance and in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. Trial activities started only after approval of the protocol was obtained from the respective sites’ Independent Ethics Committees and the Mexican National Health Authority (approval #163300410A0152, dated December 7, 2016). Written and witnessed informed consent was obtained before any study-related procedures were performed.
Sample Size Rationale
Faced with clear evidence of efficacy for the standard treatment controls, the superiority of the FDC 50/50 vs either individual component, T50 or D50, was considered. And given that the FDC 25/25 contained half the dose of each of its components, a noninferiority hypothesis was chosen for this investigational product, circumventing the use of placebo in the evaluation of a new treatment for the same condition [25]. It was estimated that a total number of 720 subjects or 180 subjects per treatment group would be required to achieve an overall power of at least 85% to reject the null hypothesis of at least one of the four formal statistical tests in the primary analysis of the trial (one-sided t test, type I error of α/4 with α = 2.5%). This assumed a common standard deviation of change from baseline values of 4 points [26, 27] and an expected treatment difference of at least 2 points [28, 29] on the primary efficacy end point (TOTPAR 4) in the comparisons between FDC 50/50 and D50 or T50, using a noninferiority margin of Δ = 1.5 points. This is half of the clinically meaningful effect of 3 points on TOTPAR4, reflecting the largest loss of effect that would still be clinically acceptable in the comparisons between FDC 25/25 and D50 or T50. To achieve 180 evaluable subjects per arm and accounting for up to 10% of subjects not being evaluable for the analysis set for the primary analysis, 200 subjects per arm or 800 subjects in total were planned to be allocated to IMPs.
Study Conduct
The trial included four visits (Figure 1), with additional unscheduled visits at the discretion of the on-site investigator. Subjects with a pain intensity score of ≥5 on an 11-point numerical rating scale (NRS) within four hours after surgery were randomized (1:1:1:1) to receive orally administered FDC 25/25 or FDC 50/50 immediate-release tablets, T50 immediate-release capsules, or D50 enteric-coated tablets. Randomization across all eight research sites was performed centrally using an interactive response voice/web system and stratified according to pain intensity at baseline (moderate [i.e., NRS score of 5 or 6] or severe [i.e., NRS score of 7-10]). Rescue medication comprising ibuprofen 400 mg (first-line therapy) and ketorolac tromethamine 30 mg intramuscular (second-line therapy) was permitted for insufficient pain relief; cold compresses or ice bags on the cheeks were allowed four hours after administration of IMPs, but not within 30 minutes before a pain assessment. Subjects were encouraged to wait at least 90 minutes after the first dose of IMP before requesting a rescue drug.
Masking Procedures
The study used double-blind and double-dummy methods to guarantee the blinding of all personnel involved in the trial. The subjects, sponsor, and investigators were blinded to the subjects’ treatments. Primary and secondary packaging of IMPs was identical for all treatment arms. Active treatments and their placebos were identical in appearance. The IMPs were packaged in a subject-specific treatment box (secondary packaging) containing three dosing kits, one kit for each dose administered (primary packaging). Each kit was marked as first, second, and third dosage and included four blisters consisting of one active treatment and three placebos to match the other three treatments. Therefore, each kit contained three tablets and one capsule of identical-appearing IMPs. Unblinding of the study was performed only after all clinical data were entered and the database was locked.
Assessments
The primary efficacy end point was total pain relief over four hours after the first dose of IMP (TOTPAR4; score range 0-16), calculated as a weighted sum of the observed pain relief scores, measured using a five-point verbal rating scale (VRS; 0 = none, 1 = a little, 2 = some, 3 = a lot, 4 = complete), over the first four hours after dose 1. Secondary efficacy end points included total pain relief over six hours and eight hours (TOTPAR6 and TOTPAR8) postdose; the summed pain intensity difference (SPID; defined as ΣPIDt × time [hours] elapsed since previous observation, where PIDt is defined as the difference between baseline pain intensity and pain intensity at time point t, and pain intensity is evaluated using an 11-point NRS ranging from 0 = no pain to 10 = pain as bad as you can imagine) between pain intensity at baseline and four, six, eight, and 24 hours postdose; time to achieve a 50% reduction in baseline pain (i.e., pain at least half gone); time to intake of first rescue medication dose; and subject’s global evaluation of the treatment (measured using a five-point Likert Scale) eight hours after dose 1 or before the first intake of rescue medication (whichever was first) and 24 hours after dose 1. Pain intensity was assessed immediately before dose 1 (baseline pain intensity) and 15, 30, 45, 60, and 90 minutes (±2 minutes) and two, three, four, five, six, seven, eight, 16, and 24 hours (±6 minutes) after dose 1. And whether baseline pain intensity was at least 50% reduced was assessed 15, 30, 45, 60, and 90 minutes (±2 minutes) and two, three, four, five, six, seven, eight, 16, and 24 hours (±6 minutes) after dose 1.
Xem thêm : The Ultimate Guide to Determining Your Child’s Hair Type(s)
Questionnaires provided to patients were in Spanish, and a verified English to Spanish translation was performed before the study.
The two-stopwatch method [30] was used to measure time to onset of first perceptible pain relief (stopwatch 1) and time to onset of meaningful pain relief (stopwatch 2).
Other exploratory efficacy end points were also considered to be analyzed in the full analysis set (FAS) population. As for the primary end point, the same analysis of covariance (ANCOVA) model was used for analyzing pain relief score after first dose over time, pain intensity score and PID after the first dose over time (based on numerical rating scale), peak pain relief score, and peak PID score. For time to request first dose of rescue medication, time to peak pain relief score, and time to peak PID, Kaplan-Meier plots, Cox hazard ratio model, and Wilcoxon log-rank test were used. Alpha levels were not adjusted for multiple comparisons.
All reported and observed adverse events were collected, as well as the start and end times, severity, any treatment required, and likely causality. The safety end points of the study were the incidence and type of AEs, which were coded to system organ class (SOC) and preferred term using the Medical Dictionary for Regulatory Activities (MedDRA). Safety and tolerability were assessed throughout the study by monitoring and evaluating treatment-emergent AEs (TEAEs), serious AEs (SAEs), severe AEs, TEAEs leading to premature discontinuation from treatment, and AEs of special interest (AESIs), including nausea, vomiting, abdominal pain, gastrointestinal bleeding (preferred term: gastrointestinal hemorrhage), dizziness, and hypotension.
Statistical Analyses
Analyses of the primary efficacy end point (TOTPAR4) and the secondary end points were performed on the full analysis set (FAS) and repeated as a sensitivity analysis on the per-protocol set (PPS). The FAS comprised all subjects allocated and treated and with at least one nonmissing pain relief assessment during the first four hours postbaseline. The PPS comprised subjects who did not use rescue medication in the first 120 minutes after the first dose, who had at least four hours of follow-up, and who complied with the protocol procedures.
The primary objective was to assess the superiority of FDC 50/50 and/or the noninferiority of FDC 25/25 vs either individual component, T50 or D50. The null hypothesis for superiority was that the mean difference (FDC 50/50 vs T50 [test 1] or FDC 50/50 vs D50 [test 2]) was ≥0, and the null hypothesis for noninferiority was that the mean difference (FDC 25/25 vs T50 [test 3] or FDC 25/25 vs D50 [test 4]) was ≥1.5. The trial was to be considered positive if at least one of the four statistical tests rejected the corresponding null hypothesis. A Bonferroni-Holm procedure was used to control the family-wise type I error rate at the prespecified one-sided significance level of α = 2.5%. The primary analysis used an ANCOVA model with treatment, site, and baseline pain intensity (assessed before IMP intake) as covariates. For the primary analysis, missing pain relief assessments in the first four hours after the first dose and pain relief assessments after start of rescue medication were imputed using the last observation carried forward with delta substitution method. Subgroup analyses were performed for the primary efficacy variable, with subjects categorized as having moderate or severe pain at baseline.
TOTPAR6, TOTPAR8, SPID, pain relief score over time, pain intensity score over time, and PID over time were analyzed with the same ANCOVA model as the primary analysis. Subjects’ global evaluations of treatment were summarized descriptively, and the secondary end points related to time to onset of analgesia and time to first use of rescue medication were presented using Kaplan-Meier plots. Medians were compared across treatment arms using the Wilcoxon log-rank test, and hazard ratios (HRs) were compared across arms using the Cox hazard ratio model. Subjects without perceptible or meaningful pain relief were censored at the end of the eight-hour period or first use of rescue medication (whichever was first).
All safety analyses were performed on the safety set, comprising all subjects allocated and treated with study medication. Safety measures were summarized descriptively and listed.
Results
Of the 1,151 subjects enrolled, 829 were randomized to treatment across the four treatment arms. Most subjects (>97%) in each treatment arm completed the full 24-hour study period (Figure 2). At least 99% of allocated subjects in each treatment arm were included in the safety set, and >88% were included in the PPS. Patients’ demographic characteristics, surgical procedures, number of teeth extracted, duration of surgery, local anesthetic dose, and baseline pain intensity characteristics were generally similar between treatment arms (Table 1). Subjects had a mean age of 23.6 years, approximately two-thirds were female, and nearly all were Hispanic or Latino of Mestizo ethnicity. Most subjects (80.3%) had all four third molars extracted. More than 93.0% of subjects from all treatment arms were compliant and received all three doses of IMPs. Approximately 41% of subjects dosed had moderate baseline pain, and 58% had severe baseline pain.
Primary Efficacy End Point
Subjects receiving FDC 25/25 (N = 204) or FDC 50/50 (N = 209) in the FAS population had higher mean TOTPAR4 scores (i.e., more pain relief) than either D50 (N = 207) or T50 (N = 205) monotherapy (least squares [LS] mean = 8.6 and 9.9 vs 5.8 and 5.4), with the greatest pain relief achieved with FDC 50/50 (Table 2). The LS mean difference in TOTPAR4 scores indicated that FDC 25/25 was noninferior (P < 0.0001) to D50 (−2.8, 95% confidence interval [CI] = −3.5 to −2.1) and T50 (−3.2, 95% CI = −3.9 to −2.5) and FDC 50/50 was superior (P < 0.0001) to D50 (−4.1, 95% CI = −4.8 to −3.4) and T50 (−4.5, 95% CI = −5.2 to −3.8) (Table 2).
A sensitivity analysis in the PPS (N = 762) and subgroup analyses in patients with moderate or severe pain at baseline showed similar findings, demonstrating the noninferiority of FDC 25/25 (all P < 0.0001) and superiority of FDC 50/50 (all P < 0.0001) to D50 and T50. All three factors assessed by ANCOVA were statistically significant: treatment (P < 0.0001), baseline pain intensity (P = 0.04), and study site (P < 0.0001).
Secondary Efficacy End Points
Pain relief assessments showed that FDC 25/25 and FDC 50/50 continued to provide greater pain relief than D50 and T50 over the eight-hour planned evaluations (Figure 3), with the LS mean differences in TOTPAR6 and TOTPAR8 assessments demonstrating the noninferiority of FDC 25/25 (P < 0.0001) and superiority of FDC 50/50 (P < 0.0001) to D50 and T50 (Table 2). Similar results were seen for SPID scores, which increased from baseline to 24 hours in all treatment groups, with greater benefits in subjects receiving FDC 25/25 or FDC 50/50 (Figure 4). The LS mean differences in SPID scores through 24 hours indicated that FDC 25/25 achieved noninferiority and FDC 50/50 achieved superiority at each time point vs either monotherapy (Table 2).
Furthermore, a 50% reduction from baseline in pain was achieved significantly (P < 0.0001) earlier with FDC 25/25 and FDC 50/50 than with D50 or T50, taking less than half the time with combination therapy than with either monotherapy (median = 1.0 and 0.8 vs 2.1 and 3.0 hours) (Table 2 and Figure 5). The mean time to intake of rescue medication was >16 hours in all treatment arms, indicating good analgesic effect in all treatment arms. Rescue medication was needed earlier by subjects in the monotherapy arms than those in the combination arms, and time to first use of rescue medication was significantly (P < 0.001) longer in the FDC 25/25 and FDC 50/50 treatment groups than in the D50 and T50 treatment groups (mean ± SD = 21.0 ± 7.5 [FDC 25/25] and 21.8 ± 6.2 hours [FDC 50/50] vs 17.4 ± 9.7 [D50] and 17.4 ± 9.8 hours [T50]) (Table 2 and Figure 6). In addition, rescue medication was also used less frequently in the FDC 25/25 and FDC 50/50 treatment groups than either D50 or T50, respectively (19.6% and 14.5% vs 35.4% and 35.1% of subjects). In terms of subjects’ global evaluation of treatment, a numerically higher percentage of subjects receiving FDC 25/25 or FDC 50/50 than those receiving D50 or T50 rated treatment as “very good” or “excellent” at eight hours (73.9% and 85.0% vs 58.7% and 52.3%) and at 24 hours (86.6% and 90.9% vs 71.4% and 68.8%) (Table 2). After three doses of FDC, it was clear that the large majority of those subjects experienced satisfactory pain relief from the FDC.
The median time to onset of first perceptible pain relief was reached more rapidly by subjects who received either combination therapy, approximately one-half the median time reported in the D50 arm and exactly one-half the median time reported in the T50 arm. Moreover, the time to onset of meaningful pain relief was also reached more rapidly by subjects who received either combination therapy. The shortest time to meaningful pain relief was in the FDC 50/50 arm (median = 1.1 hours). Moreover, the time to meaningful pain relief in both FDC treatment arms was at least half the time reported in the D50 and T50 arms (median = 1.5 and 1.1 hours for FDC 25/25 and FDC 50/50 vs 3.0 and 3.5 hours for D50 and T50, respectively). The T50 arm had the longest median time to onset of meaningful pain relief. The onset time between perceptible pain relief and meaningful pain relief (difference between the two parameters) was also shorter with combination therapy, suggesting that the improvement of relief was more rapid with the FDC at both dose levels, than the individual components.
Xem thêm : ¿Cuál es la mejor melatonina para dormir?
All exploratory end points of scores of pain relief, pain intensity, PID, peak and time to peak PID, peak and time to peak pain relief, and time to request first rescue medication favored combination therapy vs monotherapy, with the best efficacy achieved with FDC 50/50. All subjects had a peak pain relief score. Least square mean differences in peak pain relief scores and in peak PID scores favored combination therapy vs monotherapy, and both peak pain relief scores and peak PID scores were achieved earlier in the investigational arms (Table 2).
Safety
The safety profile of FDC 25/25 and FDC 50/50 was consistent with the known safety profile of the individual components, D50 and T50, without identification of unexpected safety findings. TEAEs, treatment-related AEs, and adverse events of special interest occurred at numerically lower incidences in subjects receiving D50 or FDC 25/25 than in those receiving T50 or FDC 50/50 (Table 3). The higher frequency of adverse events is consistent with exposure to higher doses of tramadol in these subjects.
The most common AEs in all treatment groups were nausea, vomiting, and dizziness, which are known adverse events of diclofenac and tramadol. Most of these events were considered related to the IMPs or rescue medication and generally occurred on day 1 before discharge from the research centers.
Across all treatment groups, severe AEs that occurred in more than one subject included vomiting (N = 6), nausea (N = 5), and dizziness (N = 3), with all nausea and vomiting events reported in subjects receiving T50 or FDC 50/50, and all dizziness events reported in those receiving FDC 50/50. One subject in the FDC 25/25 group had a TEAE (convulsion) that was considered severe and possibly related to the study drug. Overall, four subjects had eight events that led to discontinuation of IMPs: one subject with three events (nausea, hyperhidrosis, and hypotension) in the T50 group, one subject with one event (vomiting) in the FDC 25/25 group, and two subjects with two events (nausea and vomiting) in the FDC 50/50 group. None of these TEAEs that led to discontinuation were serious. No adverse events in any treatment group resulted in death.
Discussion
The results of this short-term phase 3 trial showed that combination therapy with oral FDC 25/25 and FDC 50/50 was more efficacious than monotherapy with either D50 or T50, with a safety profile consistent with the known safety profiles of the individual components. For the primary end point (TOTPAR4 scores), the FDC 50/50 group was superior and the FDC 25/25 group was noninferior to both D50 and T50 monotherapies. A subsequent post hoc analysis also demonstrated the superiority of FDC 25/25 to the monotherapies. Secondary end points also consistently showed the superiority of FDC 50/50 and the noninferiority/superiority of FDC 25/25 relative to D50 and T50, supporting the efficacy of combination therapy. Moreover, the time to onset of first perceptible pain relief and the time to onset of meaningful pain relief were reached more rapidly by subjects who received either FDC 25/25 or FDC 50/50 compared with monotherapy, and time to first use of rescue medication was significantly longer with combination therapy.
As in our study, previous studies assessing combination therapy with tramadol and diclofenac in women undergoing cesarean delivery have also shown significant reduction in pain intensity with combination therapy [17, 18]. However, when comparing combination therapy with the individual components in one study, a significant difference in the reduction in pain intensity was seen only between the combination therapy and tramadol groups [18]. It should be noted that this study differed from ours in that tramadol 100 mg and diclofenac 75 mg were administered intramuscularly [18].
In addition, two studies have compared tramadol and diclofenac combination therapy, at different dosages and/or administration routes, with tramadol and acetaminophen (paracetamol) combination therapy. In one study in women undergoing cesarean delivery, intravenous (IV) tramadol (75 mg six-hourly) plus rectal diclofenac (100 mg eight-hourly for 24 hours) combination therapy reduced the overall pain score to a greater extent than tramadol plus IV acetaminophen (1 g six-hourly) combination therapy [19]. Consistent results were obtained from a second study, which showed that a tramadol (50 mg)/diclofenac (75 mg) FDC (one tablet twice daily) reduced pain intensity to a greater extent than a tramadol (37.5 mg)/acetaminophen (325 mg) FDC (two tablets every four to six hours, up to a maximum of eight tablets daily) in patients with postoperative pain, acute musculoskeletal conditions, or acute flare of osteoarthritis and rheumatoid arthritis [31], further supporting the efficacy of tramadol/diclofenac FDC.
Due to the opioid epidemic in the United States, and based on currently available evidence, the American Dental Association advocates that dentists should prescribe NSAIDs alone or in combination with acetaminophen over the opioid class as first-line therapy for acute pain management [32]. Our study clearly demonstrates that tramadol/diclofenac is superior when compared with an NSAID (diclofenac) or tramadol alone in the setting of third molar tooth extraction, with no new safety signals. The setting used in this study was chosen as a well-established pain model to illustrate and test the relative efficacy and safety of the investigational FDCs compared with the corresponding monotherapies. These results may apply as alternatives for dentists to treat patients after third molar extraction, but are not restricted to this pain indication. This study indicates that there is a significant dose response in the efficacy and side effect frequency of this FDC. A general principle in pain therapeutics is to prescribe the lowest effective dose of a drug for any particular patient. Patients experiencing mild pain, such as after a non-bony dental surgical procedure or extraction or other similar conditions, would likely benefit from the lowest dose of this combination. However, patients who have undergone bony surgery or other types of major surgery or tissue injuries and who are expected to have moderate to severe pain for several days would likely benefit most from the higher dose during the first few days.
It is also important to stress that the current study assessed the short-term use (24 hours) of the tramadol/diclofenac combination for the management of acute dental pain. Although tramadol and its metabolites also interact with µ-opioid receptors and long-term use can be associated with opioid-like adverse outcomes (e.g., abuse, dependence, tolerance) [14], the safety profile of the tramadol/diclofenac combination after short-term use was consistent with the known safety profile of the individual components.
As our study was conducted in a relatively healthy, young population in a controlled clinical setting, the role of comorbidities and concomitant medications could not be assessed, which should be considered a limitation of this study. However, our findings suggest that other types of moderate to severe acute pain, such as that experienced after orthopedic surgery or major thoracic surgery, may benefit from similar short-term multimodal therapy.
Conclusions
In summary, the results of our short-term study indicate that tramadol/diclofenac FDC 25/25 and FDC 50/50 combination therapies are complementary and provide superior analgesia relative to T50 and D50 monotherapies after third molar extraction. The safety profile of the fixed-dose combinations is consistent with the known safety profile of the individual components.
Funding sources: Grünenthal S.A. sponsored the trial.
Conflicts of interest: Dr. Desjardins was a paid consultant to Grünenthal S.A. and the coordinating investigator for this study. All other authors were principal investigators at participating trial sites and received honoraria for their roles as principal investigators. None of the authors received any payment for their contributions to the publication of this paper.
Acknowledgments
Paul Desjardins, DMD, PhD, was the coordinating investigator, and his colleagues Peter Black and Erin Conte coordinated the translation of the pain questionnaires and trained the study staff in pain assessment techniques. Special thanks to the following investigators, who participated and enrolled subjects in Mexico: Juan Eduardo Arizpe Coronado, DDS, San Luis Potosí; José de Jesús Martín Barrera Arellano, DDS, Chihuahua; Gabriela Margarita Avalos González, DDS, Zapopan; Martha Gil Almuina, DDS, Monterrey; Rogelio Gabriel Guajardo Rodriguez, MD, León; Manuel de Jesús González De Santiago, DDS, Aguascalientes; Fabian Alvarado Villanueva, DDS, Puebla. The authors also acknowledge the Grünenthal scientists who participated in the study design and analysis plan and staff from ReSolution Latin America Clinical Research Solutions (Buenos Aires, Argentina) and InClin, Inc. (San Mateo, CA, USA), who assisted in monitoring, data management, drug distribution, and statistical analysis. Medical writing support, under the guidance of the authors, was provided by David P. Figgitt, PhD, ISMPP, CMPP, and Sohita Dhillon, PhD, Content Ed Net, with funding from Grünenthal S.A.
References
Nguồn: https://blogtinhoc.edu.vn
Danh mục: Info
This post was last modified on Tháng mười một 19, 2024 5:06 chiều